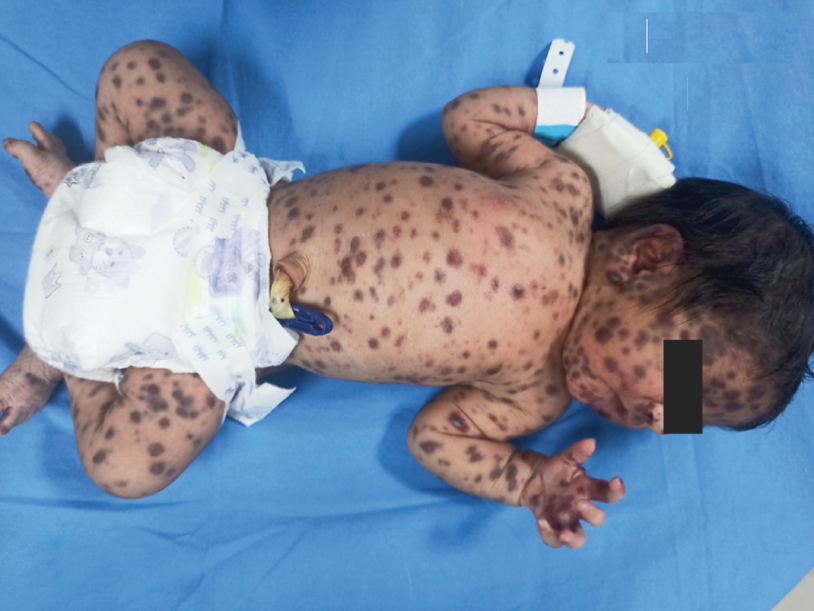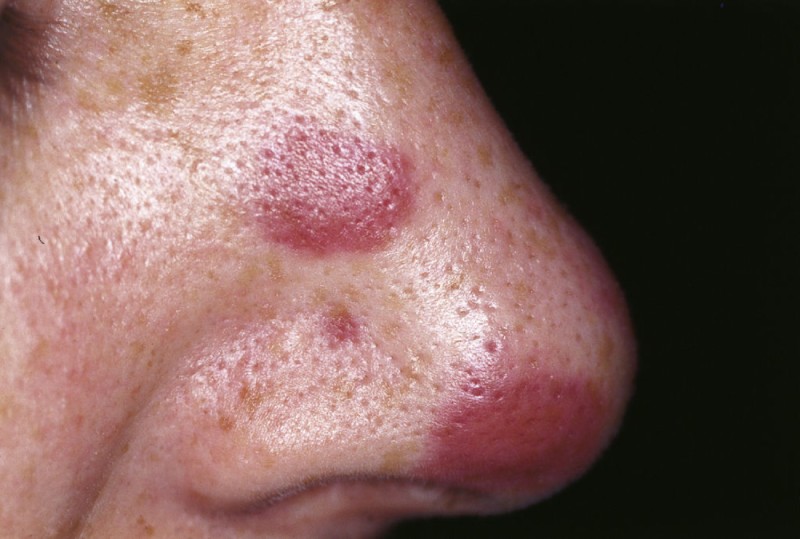
History: A 33 yr female presented with fatigue & flat painless purple lesions on the nose. She had a history of HIV infection and a low CD4 cell count. What’s the diagnosis?
1. Hemangioma
2. Kaposi Sarcoma
3. Bacillary Angiomatosis
4. Purpura
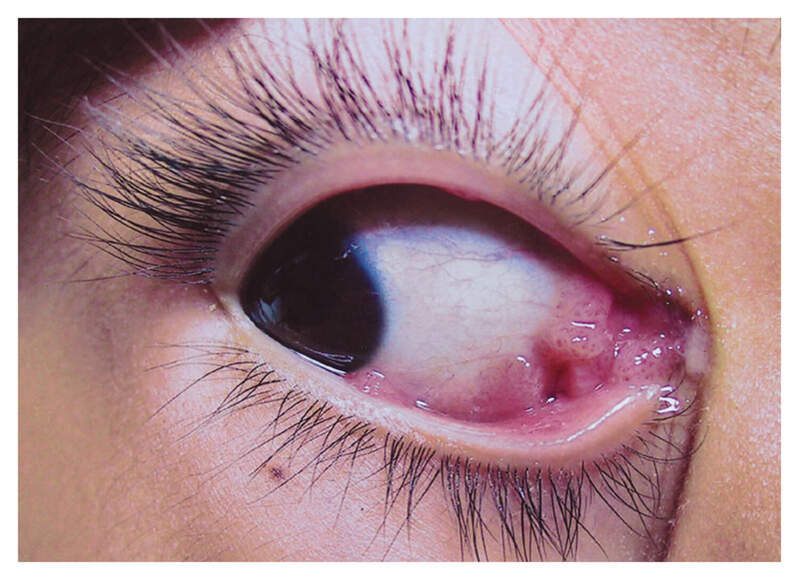
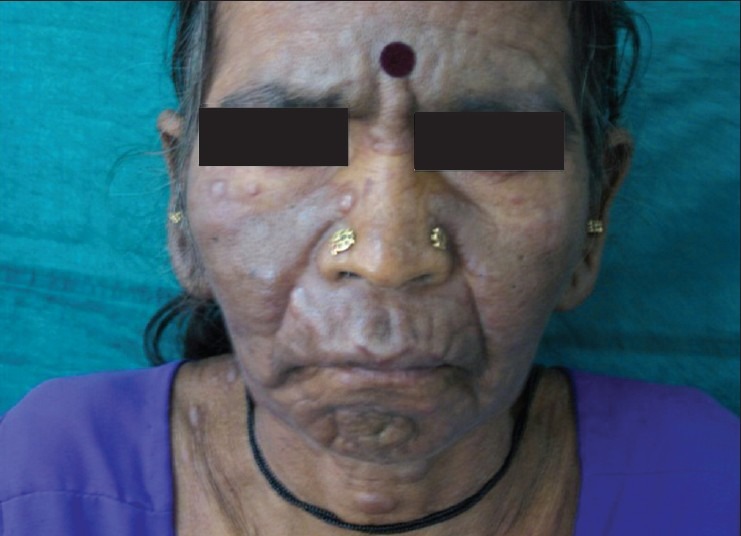
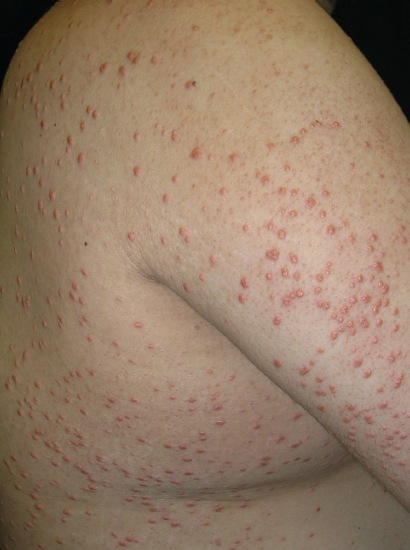
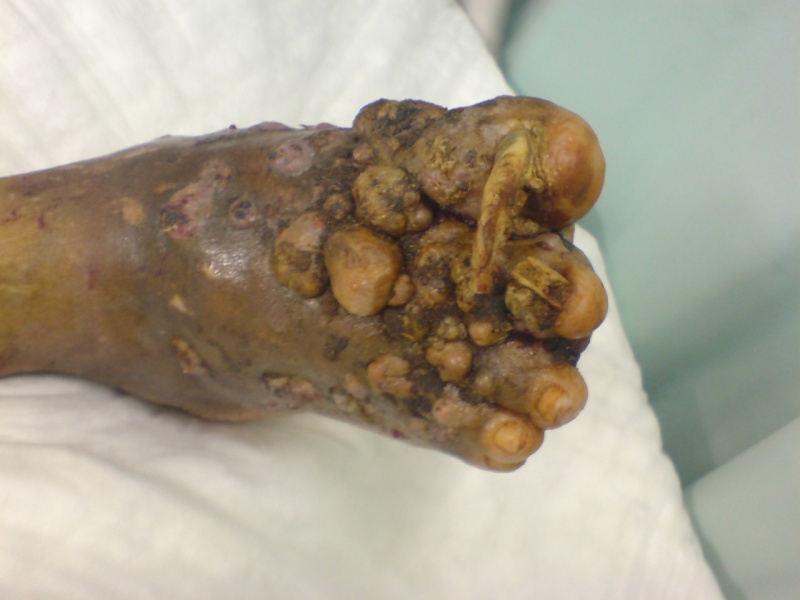
Answer: Kaposi sarcoma (KS) is an indolent angio-proliferative spindle-cell tumor derived from endothelial and immune cells infected with human herpes virus type 8 (HHV-8).
Four sub-types are described:
- Classic KS tends to affect older men in regions where KSHV is highly prevalent (Mediterranean, Eastern Europe, Middle East), is usually slow-growing, and most often affects only the legs.
- Endemic KS is MC in Sub-Saharan Africa and is more aggressive in children, while older adults present similarly to classic KS.
- Immunosuppression therapy-related KS generally occurs in people following organ transplantation and mostly affects the skin.
- Epidemic KS occurs in people with AIDS and many parts of the body can be affected.
Symptoms: Lesions are nodules or blotches that may be red, purple, brown, or black, and are usually papular. They are typically found on the skin, mouth, gastrointestinal tract, and respiratory tract. Growth can range from very slow to explosively fast, and is associated with significant mortality and morbidity. The lesions are painless, but become cosmetically disfiguring or interruptive to organs.
Diagnosis: KS is diagnosed by tissue biopsy, CD4 count
Treatment: HAART, Combination therapy with regimens such as ABV (actinomycin D, bleomycin, vincristine) or single-agent therapy such as doxorubicin. Cytotoxic agents like doxorubicin, daunorubicin, Paclitaxel or oral etoposide. Radiation therapy, cryotherapy, laser therapy or surgical excision.